Analysis of S-S Bonds
If the gene sequence that codes for the target antibody indicates a cysteine residue, the Guidelines require the determination
of the number and positions of all free sulfhydryl groups and disulfide bonds. This is evaluated using peptide mapping (under reducing conditions and non-reducing conditions), mass spectrometry or another suitable method.
Introduced here is the analysis using a combination of peptide mapping and mass spectrometry. This method involves digesting the target protein in a proteolytic enzyme such as trypsin under various conditions and comparing the molecular weights of the generated peptide fragments with the theoretical values. The test procedures and results for the analysis of human lysozyme are introduced.
Experiment and research flow
- Use the cell-free protein synthesis kit (Transdirect insect cell) to synthesize human lysozyme in a test tube. (Fig. 1 shows the sequence of the synthesized human lysozyme.)
- Purify in an affinity column (Strep-Tactin super flow, manufactured by QIAGEN, Germany).
- Denature using 8M urea.
- Trypsin digestion after three types of pretreatment: untreated (retaining S-S bond); S-alkylation (alkylation of any free SH groups that exist); reduction and alkylation (reduction of S-S bonds followed by alkylation of SH groups)
Results
Fig. 1 shows the amino acid sequence and predicted S-S bonds (indicated by straight lines) of the human lysozyme synthesized in the test tube. Table 1 shows the theoretical values and MS measured values for the trypsin-digested peptide fragments (associated with S-S bonds).
Fig. 2 shows part of the MS spectrum of the trypsin-digested peptide fragments.
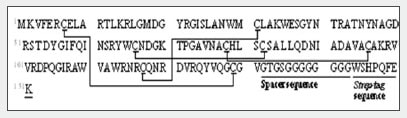
Fig. 1 Amino Acid Sequence and Predicted S-S Bonds of the Synthesized Human Lysozyme
The m/z 3239.22 ion disappears due to reduction and alkylation of peptide fragment B, indicating that peptide 3 and peptide 4 form intramolecular S-S bonds (Fig. 2). The same method confirmed intramolecular S-S bonds in peptide fragment C. (Data omitted.)
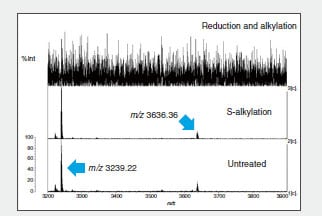
Fig. 2 MS Spectrum of Recovered Peptide Fragments (m/z 3200 to m/z 3900)
Comparison of the reduction/alkylation and untreated MS
spectra for peptide fragment A indicates that peptide 1 and
peptide 2 form intramolecular S-S bonds. However, the
combination with the cysteine residue cannot be determined
from the results in Fig. 2 only.
To confirm this combination, the peptide 1 intramolecular
S-S bonds were identified using LC to separate and purify
peptide fragment A, digestion in another protease
(thermolysin), and determination of the mass of the
generated fragments (Fig. 3).
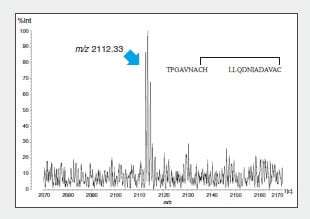
Fig. 3 MS Spectrum of Thermolysin Digest of Peptide Fragment A (m/z 3636.36)
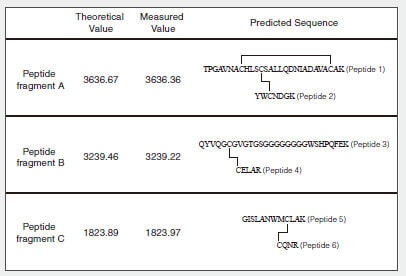
Table 1 Theoretical Values and Measured Values for Predicted Peptide Fragments (Associated with S-S Bonds)
AXIMA Performance MALDI-TOF MS

The AXIMA Performance is one of the most powerful tools in mass spectrometry, delivering information-rich spectra with greater sensitivity and higher confidence in identification. It is an extremely versatile and powerful TOF-TOF system, integrating workflows for a diverse range of analytical needs.
- From high energy MS/MS of proteomics and other biological and organic samples to uncompromised analysis of high mass intact proteins
- True high energy MS/MS - CID with a laboratory frame collision energy of 20keV
- Optimal precursor ion selection resolution using revolutionary gating technology
- Outstanding sensitivity - uncompromised design, to ensure no MS/MS signal is discarded