Rapid Isolation of Peptides and Proteins from Biological Fluids for Proteomic Analysis by MALDI-TOF Mass Spectrometry
-
- Proteomic profiling by matrix assisted laser desorption/ionization (MALDI) mass spectrometry (MS) has been shown to be effective at assessing systemic disease
- A method to rapidly isolate and concentrate peptides and proteins in biological fluids within a chosen mass range
- By characterizing these changes in the plasma and serum, insight can be gained into the biochemical events that underpin various disease states.
-

MALDI-TOF mass spectrometry is a versatile analytical technique capable of rapidly characterizing biological samples. The ease of use and high throughput capability make this approach advantageous for analyzing large sample sets. In comparison, the time of data acquisition is significantly less than the initial sample preparation. The isolation, concentration, and buffer exchange necessary for sample preparation can be costly and time consuming. Therefore, new methods need to be developed to isolate and concentrate target molecules quickly and efficiently. In this study, we developed a method to rapidly isolate and concentrate peptides and proteins in biological fluids within a chosen mass range. This method was then applied to mouse plasma, and serum to define a healthy baseline.
Proteomic profiling by matrix assisted laser desorption/ionization (MALDI) mass spectrometry (MS) has been shown to be effective at assessing systemic disease1-4, . Other targeted detection technologies such as competitive immunoassays, sandwich-type immunoassays, immune-capture, immunoblots, and activity assays have traditionally been employed to diagnose various disease states. However, MALDI-TOF MS is a sensitive, high throughput, and cost-effective technique that is well suited for analyzing biological fluids. Changes in the relative amounts of peptides and proteins in plasma reflect systemic events; therefore, they can be measured and serve as diagnostic, predictive or prognostic biomarkers of disease. By characterizing these changes in the plasma and serum, insight can be gained into the biochemical events that underpin various disease states.
Methods
A standard mixture of peptides and proteins was processed by dual membrane filtration to ensure the proper target molecules were isolated. Mouse plasma and serum samples for this study were supplied by the Shimadzu Institute of Research Technologies Animal Care Facility at University of Texas, Arlington with permission following IUCUC approved experiments. A non-homologous internal standard was spiked into all biological samples. Peptides and proteins were isolated from each biological fluid and analyzed by MALDI-TOF mass spectrometry. For all mouse samples multiple biological and technical replicates were spotted with sinapinic acid for analysis. All mass spectra were acquired in linear and positive ion mode within a mass range of 1-80kDa.
Peptide and Protein Standards
- Glu-Fibrinopeptide, Cytochrome C, Myoglobin, and Bovine Serum Albumin were prepared at a concentration of 5 pmol/ul in 70/30 deionized water and acetonitrile with 0.1% Trifluoroacetic acid.
- Using the dual membrane device (Figure 1) the peptides and proteins were separate into three fractions; less than 1kDa (Low), less than 50kDa (Mid), and greater than 50kDa (Top).
- All peptide and protein standards were spotted 1:1 ratio with 40mg/ml of Sinapinic acid (SA) and analyzed by MALDI-TOF MS in linear mode.
Plasma and Serum Samples
- Approximately 500ml of plasma and serum was extracted from each mouse.
- Prior to processing the plasma and serum samples were diluted 1:20 in 1ml of [30% / 70% / 0.1%] acetonitrile / dH2O / trifluoroacetic acid and an internal standard was added Glu-Fibrinopeptide at a concentration of 500 fmol/µl.
- Relative quantification was performed using internal standard / analyte peak area ratios.
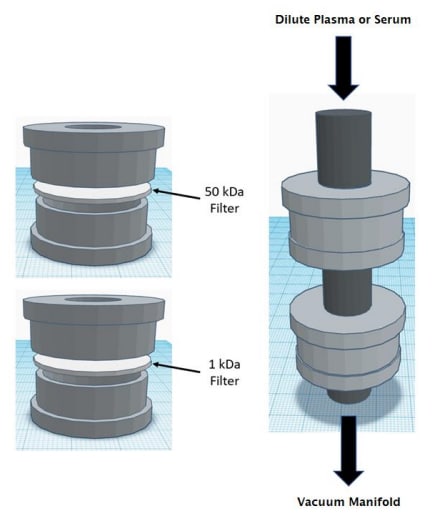
Figure 1: Dual membrane filtration device
We 3D printed a dual filtration system to isolate peptide and protein molecules from biological samples. The system was designed to use molecular weight cutoff filters or dialysis membranes for separation. Initially, we tested the apparatus with a mixture containing one peptide and three proteins (Glu-Fibrinopeptide, Cytochrome C, Myoglobin, and Bovine Serum Albumin). MALDI-TOF MS was used to acquire spectra from the low, mid, and high fractions Figure 2. These data show we were successful at isolating Cytochrome C and Myoglobin. However, the Bovine Serum Albumin was not completely removed from the sample. Additionally, it was observed that Glu-Fibrinopeptide was present in the middle fraction but a small amount was not capture by the low molecular weigh cutoff filter. There are three possible reasons for this outcome. First, the buffer system used to dilute the sample could affect the filters porosity. Second, it is conceivable a leak could cause molecules to circumvent the filters. Lastly, the structure of the molecules could allow them to pass through the pores. To overcome these potential pitfalls, we can alter the design of the filtration system and evaluate new molecular weight cutoff filters or dialysis membranes.
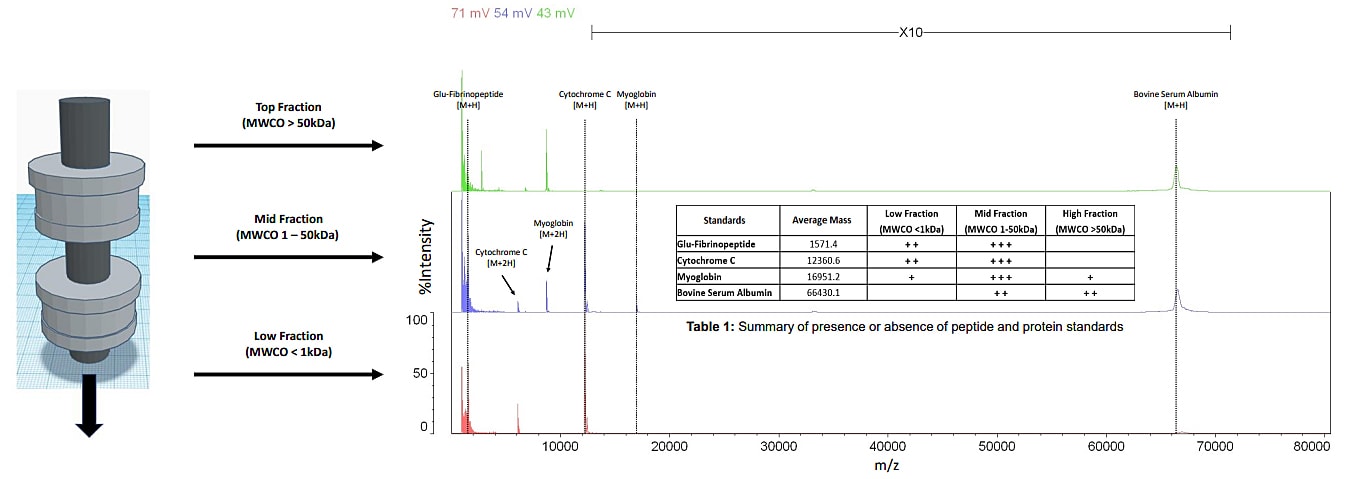
Figure 2: A mass spectrum of peptide and protein standards analyzed by MALDI-TOF MS after
dual filtration (mass range 1 - 80 kDa)
In addition to peptide and protein standards, we ran mouse plasma and serum through the dual filtration device. We wanted to observe how reproducible our data was when using the device to process a complex mixture of molecules. Figure 3 and Figure 4 display mass spectra of the mid fraction from the plasma and serum samples. Across the 1 - 80kDa mass range five peaks were used to test reproducibility. The peak area ratio of internal standard to analyte was calculated. Multiple technical replicates were acquired to determine the mean, standard deviation, and coefficient of variance. The coefficient of variance for both the plasma and serum samples ranged from 10%-25% (data not shown). This higher than normal amount of variability is the result of inconsistent retention of molecules in solution. To address this issue, different molecular weight cutoff filter materials and mass ranges will be tested.
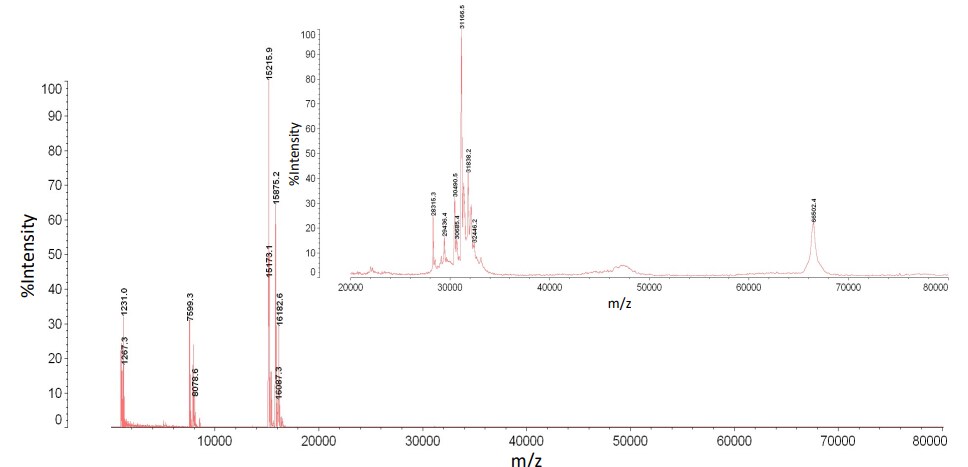
Figure 3: A mass spectrum of mouse plasma analyzed
by MALDI-TOF MS after dual filtration (mass range 1 - 80 kDa)
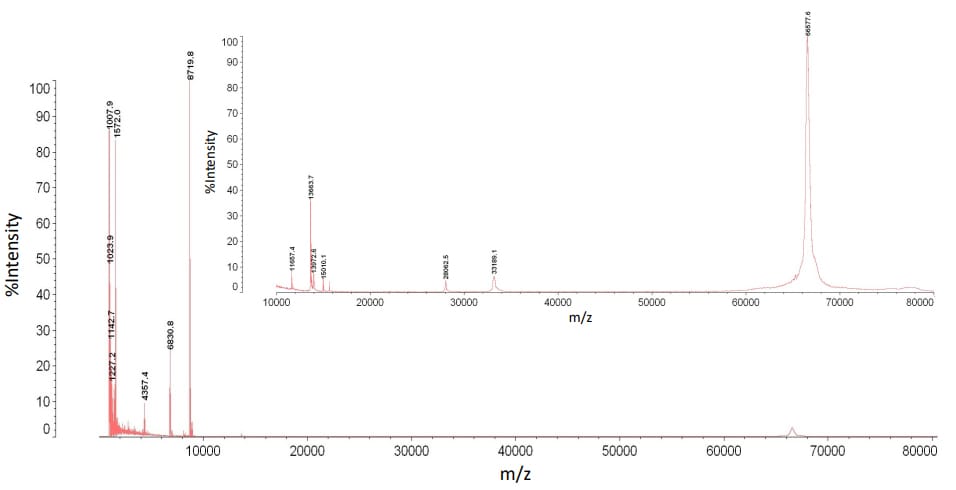
Figure 4: A mass spectrum of mouse serum analyzed
by MALDI-TOF MS after dual filtration (mass range 1 - 80 kDa)
Conclusion
It is well established that MALDI-TOF MS is a versatile analytical technique that is high throughput, easy to use, and a cost-effective way to analyze biological fluids. However, the lag time that arises from sample prep needs to be minimized. The ability to rapidly prepare samples for MALDI-TOF analysis is vital for biomarker discovery. Therefore, new methods need to be developed to screen molecules in biological fluids. To thoroughly survey all the molecules present in plasma and serum there are a few considerations to take into account. The removal of small molecular weight contaminants and high molecular weight proteins can help to minimize signal suppression. Moreover, buffer exchange and concentration can help to enhance the signal of molecules in low abundance. The development of this device was meant to address the aforementioned consideration. However, further optimization and testing will have to be performed to improve the device and to allow for routine use.
References
- Swiatly, A. Horata, A. Hajduk, J. Matysiak J. Nowak-Markwitz, E. and Kokot, Z. J. (2017). MALDI-TOF-MS analysis in discovery and identification of serum proteomic patterns of ovarian cancer. BMC Cancer 17: 472
- Pietrowska, M. and Widiak P. (2012). MALDI-MS-Based Profiling of Serum Proteome: Detection of Changes Related to Progression of Cancer and Response to Anticancer Treatment. Int. J. Proteomics 2012: 926427.
- Böhm, D. Keller, K. Pieter, J. Boehm, N. Wolters, D. Siggelkow, W. Lebrecht, A. Schmidt, M. Kölbl, H. Pfeiffer, N. and Grus, F. H. (2012). Comparison of tear protein levels in breast cancer patients and healthy controls using a de novo proteomic approach. Oncol Rep. 28(2):429-38.
- Lebrecht, A. Boehm, D. Schmidt, M. Koelbl, H. Schwirz, R. L. and Grus, F. H. (2009). Diagnosis of Breast Cancer by Tear Proteomic Pattern. Cancer Genomics Proteomics. 6(3):177-82.
MALDI-TOF Mass Spectrometry

- Employs unique quadrupole ion trap technology for highly sensitive and accurate MSn spectral measurements of the molecular ions produced by MALDI.
- The high precursor ion selectivity offers reliable selection of the target ions only, even in the presence of large numbers of impurity ions, to permit acquisition of high-quality MSn analysis data.
- Offers highly accurate MSn analysis data that allows accurate structural prediction.
- Analysis software provides powerful support for glycopeptide structural analysis.