Post-Translational Modification Analysis | Phosphorylation Analysis by MALDI-TOF MS (2)
Sequence Analysis of Phosphopeptides by Seamless PSD
Recently, mass spectrometry has been applied to the analysis of protein phosphorylation and phosphorylation sites. The phosphorylation sites are determined by a variety of types of MS/MS.
A method of analyzing the phosphorylation modification sites using seamless PSD (sPSD) with the MALDI method is described below.
Collision-induced dissociation (CID) is commonly used for amino acid sequence analysis by MALDI-MS/MS. However, due to the dissociation of the fragile phosphate groups caused by collisions with the inert gas, it is difficult to obtain information on phosphorylation sites using this method.
MS/MS measurements using seamless PSD (sPSD) require no inert gas and can obtain MS/MS spectra with some suppression of phosphate group dissociation.
Fig. 1 shows the seamless PSD (sPSD) spectrum of a doubly phosphorylated peptide obtained using DHB as the matrix. From the fragment ion assignment results in Fig. 1, it is apparent that y-ions, which are the C-terminal fragments, are formed preferentially.
While phosphate group dissociation from the fragment ions was observed, the intensity is low (y7-98 in diagram). The MS/MS spectrum obtained is adequate to decode the entire sequence from the masses of the phosphorylated amino acid residues.
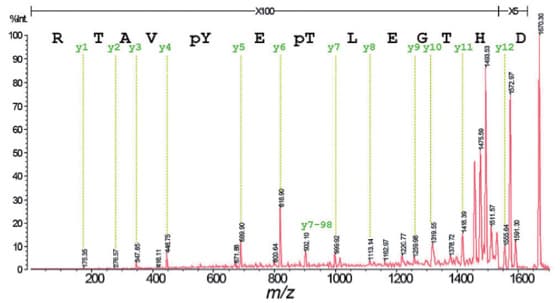
Fig. 1 sPSD Spectrum of MAP Kinase Fragment 177-189 (a doubly-phosphorylated peptide)
This effect is known to be amplified when DHB is used, but conventional PSD was not practical due to insufficient fragment ion detection sensitivity. A major feature of sPSD is its superior fragment ion generation efficiency to PSD. This practical level of sensitivity permits MS/MS measurements of phosphopeptides.
AXIMA Performance MALDI-TOF MS

The AXIMA Performance is one of the most powerful tools in mass spectrometry, delivering information-rich spectra with greater sensitivity and higher confidence in identification. It is an extremely versatile and powerful TOF-TOF system, integrating workflows for a diverse range of analytical needs.
- From high energy MS/MS of proteomics and other biological and organic samples to uncompromised analysis of high mass intact proteins
- True high energy MS/MS - CID with a laboratory frame collision energy of 20keV
- Optimal precursor ion selection resolution using revolutionary gating technology
- Outstanding sensitivity - uncompromised design, to ensure no MS/MS signal is discarded