Extraction of N-terminally Blocked Proteins from Electrophoresis Gels and Sequence Analysis Using a Benchtop MALDI-TOF Mass Spectrometer
- Combining protein extraction from electrophoresis gels and the MALDI-8020 benchtop MALDI-TOF mass spectrometer to measure molecular weights of N-terminally blocked proteins and perform sequence analysis by ISD
- Carbonic anhydrase extracted from the protein gel was mixed with sinapic acid or DAN, analyzed in linear mode using the MALDI-8020
- Using "Mass++™" and Basic Local Alignment Search Tool (BLAST), a homology search for the amino sequence obtained from the detected peaks
Proteins may now be easily identified using trypsin digestion pretreatment and a mass spectrometer. This type of workflow is well-established and is accessible even to non-MS experts. In such workflows, protein identification is achieved through simple database searching with a search engine, such as Mascot (Matrix Science). However, in the case of terminal sequence analysis of proteins that have undergone processing or mass spectrometric identification of proteins from minor biological species not listed in databases, more expensive high-end instrumentation and more complex workflows, requiring skilled operators, are often employed. Furthermore, protein sequencers are generally used as a method for protein terminal sequence analysis; but in the case of N-terminally blocked proteins, de-blocking is necessary as a pretreatment to perform analysis. The pretreatment may not work well depending on the protein type and therefore requires a level of skill/experience which may limit its use.

In recent years, it has become possible to analyze the sequence of proteins whose N-terminal is blocked or which are not registered in databases by utilizing protein fragmentation that occurs in the ion source (ISD: In-Source Decay) of MALDI-TOF mass spectrometers. In addition, ISD is theoretically not restricted by the mass of each sample, so high-mass proteins can be sequenced directly without trypsin digestion.
Nevertheless, since ISD requires the target protein to be isolated and purified, protein sequence analysis by ISD has yet to gain popularity with non-experts of mass spectrometry.
As a solution to such problems, this article introduces an example of combining protein extraction from electrophoresis gels and the MALDI-8020 benchtop MALDI-TOF mass spectrometer (Fig. 1) to measure molecular weights of N-terminally blocked proteins and perform sequence analysis by ISD.
Materials and Methods
Bovine carbonic anhydrase (Sigma-Aldrich), whose N-terminal is acetylated, was used as a model sample. The sample was dissolved in a buffer solution for electrophoresis, heated at 95 °C for 5 minutes, and electrophoresed on a polyacrylamide gel (ATTO 12.5 %, precast e-PAGEL). The resulting polyacrylamide gel was stained with CBB (ATTO EzStain AQua) to detect protein spots (Fig. 2).
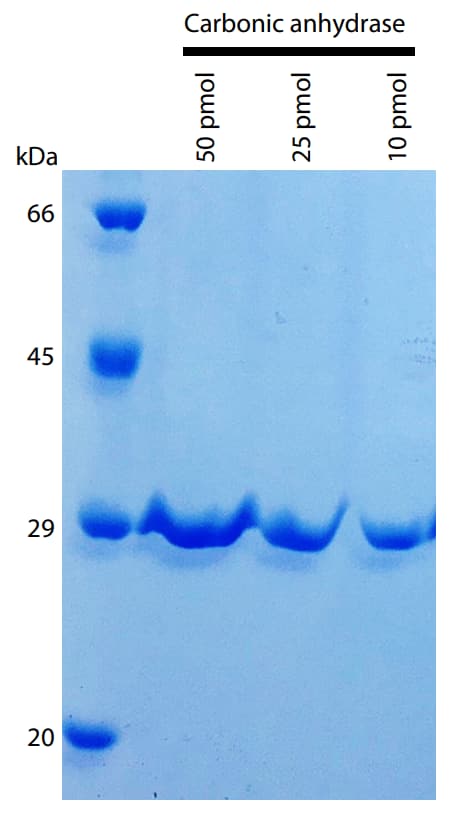
Fig. 2 Electrophoresis Image of Carbonic Anhydrase
Using an extraction buffer solution containing surfactant, we extracted proteins from the bands of the gel-separated carbonic anhydrase. We then precipitated the proteins in the extraction buffer solution using chloroform/methanol to remove surfactant and salts and conducted measurements using the MALDI-TOF mass spectrometer. Sinapic acid was used as MALDI matrix for protein molecular weight measurement and 1,5-diaminonaphthalene (DAN) for sequence analysis by ISD.
Results
The carbonic anhydrase extracted from the gel was mixed with sinapic acid and was analyzed in linear mode using the MALDI-8020. The result is shown in Fig. 3. A peak corresponding to the singly-charged protonated carbonic anhydrase extracted from the 10 pmol protein spot/band was detected near m/z 29030.
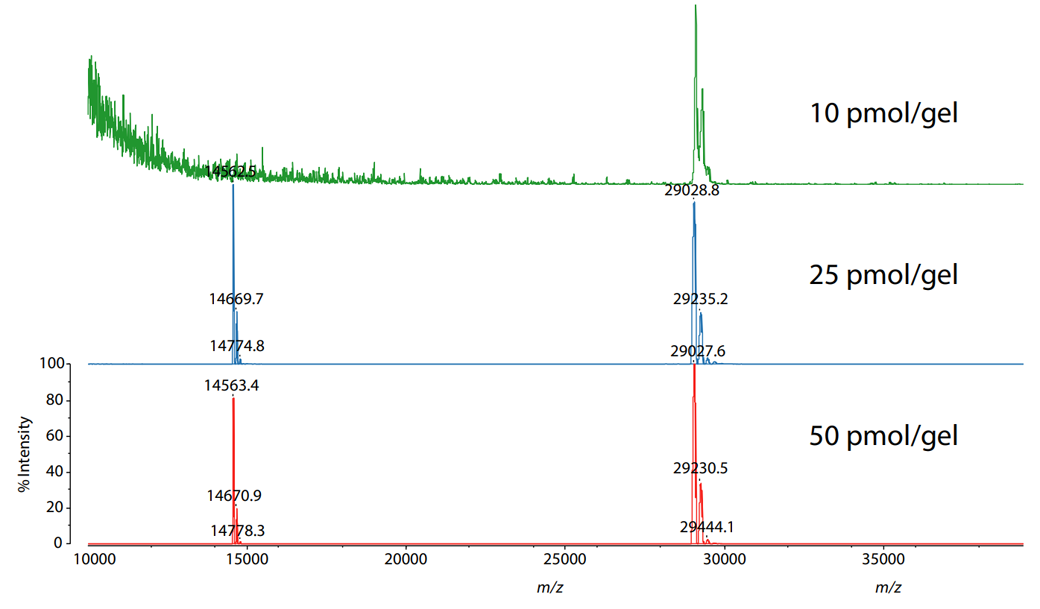
Mass Spectra of Carbonic Anhydrase Extracted from Polyacrylamide Gels
Next, the carbonic anhydrase extracted from the 25 pmol protein band was mixed with DAN and analyzed in linear mode, again using the MALDI-8020. The result is shown in Fig. 4. Peaks consistent with the masses of c-ions (N-terminal fragments generated from a protein by ISD) were predominantly detected (center in Fig. 4, bottom in Fig. 4). By using free software "Mass++ ™" and Basic Local Alignment Search Tool (BLAST), which is an amino acid sequence alignment tool for proteins, we conducted a homology search for the amino sequence obtained from the detected peaks. This resulted in carbonic anhydrase being matched as the highest probability (bottom right in Fig. 4). In addition, from the detected c-ion fragment masses and the amino acid sequence of carbonic anhydrase registered in the databases, we can infer that the N-terminal sequence is SHHWGYGKH... and is Nacetylated (top in Fig. 4).
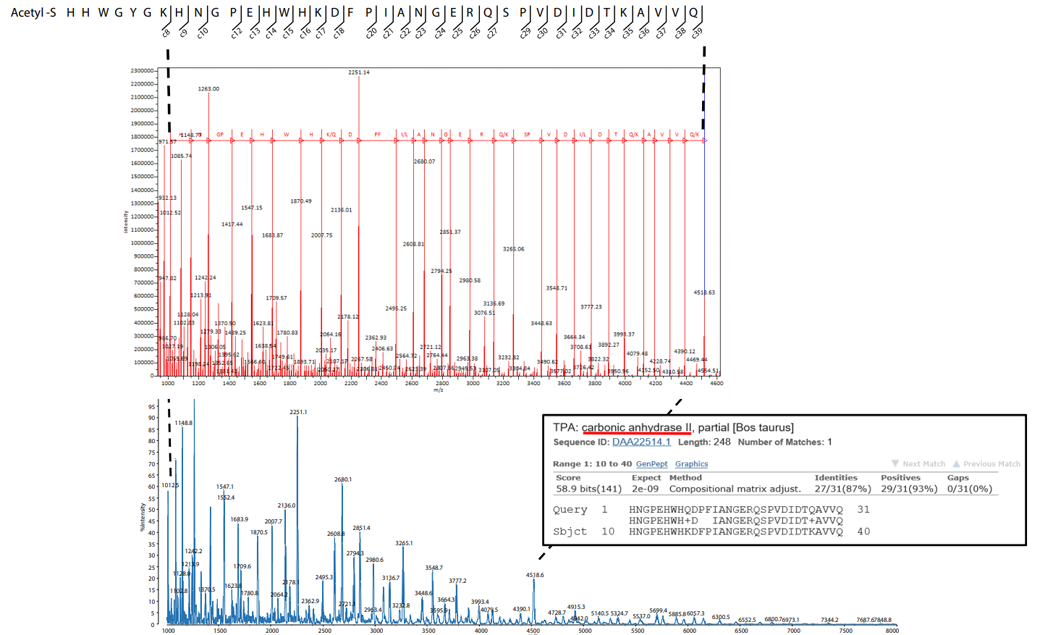
Fig. 4 MALDI-ISD Analysis Results of Carbonic Anhydrase Extracted from Polyacrylamide Gels Top: Inferred Amino Acid Sequence, Center: Result of Identification with Mass++, Bottom: ISD Spectrum, Inset: BLAST Search Result
Conclusion
Following extraction of proteins from electrophoresis gels, we measured the molecular weight of N-terminally blocked proteins and, subsequently, performed sequence analysis by ISD using the MALDI-8020 benchtop MALDI-TOF mass spectrometer. The results suggest that (partial) sequence information of proteins whose exact sequence is not registered in databases can still be determined and that the terminal sequence of proteins that have undergone processing can also be analyzed using this technique. Of the proteins extracted from electrophoresis gels, the minimum concentration which permitted sequence analysis of proteins by ISD was 25 pmol/gel. Higher sensitivity or more efficient protein extraction is required for the analysis of lower protein amounts by this technique. However, there is a case report1 of sequence analysis using a mass spectrometer by collecting proteins from dissolvable polyacrylamide gels at a high yield rate. Future development can be expected.